Measuring Chronological Age
Aging is something that we all experience, but that we still struggle to understand. We know that aging is natural and allows us to mature, and is ultimately related to natural mortality. Many researchers have sought to develop a measure for aging in our cells. A measure of aging can help us to understand if certain events and behaviors make us age faster, like smoking or childhood trauma. Some people also believe that, by understanding why we age, we might have a chance at reversing it and finally finding that fountain of youth. There are major ethical concerns as we enter this uncharted territory, but the study of chronological age provides many new avenues for research. At UCLA, Dr. Steven Horvath has developed a novel and incredibly accurate method for measuring chronological age.
According to the American Federation for Aging Research (AFAR), a biomarker of aging must meet the following criteria: it must predict the rate of chronological aging (aka how old you are); monitor a basic process involved in aging, not the effects of a disease; be able to be tested repeatedly without harming the person; and work in humans as well as in laboratory animals (Horvath, 2013). You may have heard of another proposed biological clock—telomeres, which are related to cancer—but epigenetics may provide an even more accurate measure of age.
Epigenetics and Genetic Drift
Figure 1. Double-stranded DNA is typically in the form of a double helix and is made up of a unique pattern of four nucleic bases (ATCG).https://s-media-cache-ak0.pinimg.com/236x/66/9d/b2/669db24d96259566dcb6d6cfea0ddc86.jpg
Before we get into the epigenetic clock, we’ll talk about genetics. Genetics refers to the study of genes, passed from parents to children, which are made up of DNA. DNA consists of unique combinations of four nucleic bases—adenine, thymine, guanine, and cytosine. Your DNA is your genetic blueprint and is really what makes you different from everyone else. All of your cells contain the same DNA, and this genetic combination determines the proteins your cells produce and contributes to why we all look, act, and think differently. All cells in the body contain the same DNA, but we know that not all of our cells are the same. You have muscle cells, neurons, and heart cells, all of which have different proteins. Different types of cells need to produce different types of proteins, and they can do this because their DNA is not exactly identical. Although the DNA sequence itself is the same in all your cells, the structure of your DNA can change. Different chemical groups can be added to your DNA and cover it so that certain parts of it are not expressed. These changes to your DNA structure are referred to as epigenetics (Singal & Ginder, 1999; Berger, Kouzarides, Shiekhattar, & Shilatifard, 2009).
The most common form of epigenetics is DNA methylation. DNA methylation refers to when a methyl group is added on to a nucleic acid, typically cytosine. Think of it this way—your DNA is wound up in a double helix with the strands bound to one another, like the strands of a zipper. In order for your genes to be read, your strands of DNA need to separate—you need to unzip the zipper in order to be able to see what is really inside. When the DNA is really tightly bound, then you can’t see anything. The parts of your cell involved in making proteins can’t see anything either, so they cannot produce proteins. Methylation makes certain parts of the DNA very tightly bound, so that the strands separate less and the proteins from those genes are not produced. This process prevents certain proteins to form, which is important since your skin cells need different proteins than your brain cells. Epigenetics is essential in allowing your cells to form different tissues. However, epigenetics may also play a role in aging.
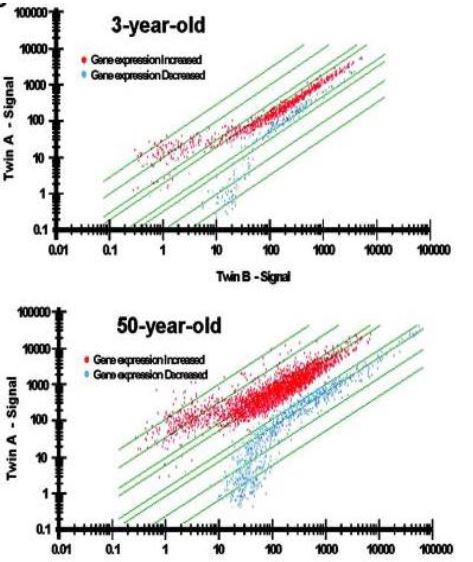
Figure 2. Epigenetic drift is greater in 50-year-old twins than in 3-year-old twins (Fraga, et al., 2005).
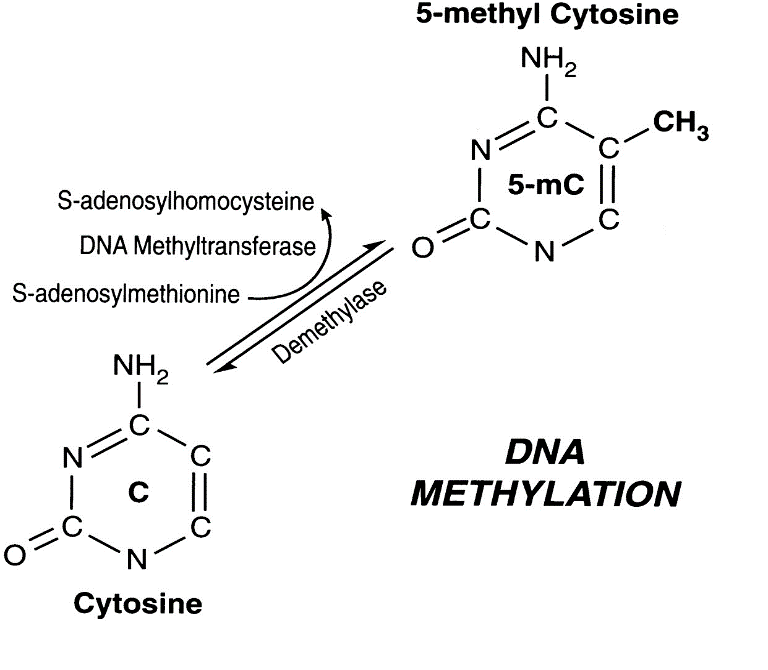
Figure 2. Interconversion between cytosine and 5-methyl cytosine (Singal & Ginder, 1999).
Epigenetic modifications clearly occur naturally as we age. A study of identical twins found that as toddlers, their DNA was mostly just that—identical. However, their DNA became gradually different over time (Fraga et al., 2005). In Fig. 2, The Y-axis represents one twin and the X-axis represents the other. A straight diagnal line across the graph suggests that the genes are expressed equally in both twins. Although there are a few 3-year-olds who show big differences in genetic signaling, most fall along the line. However, among the 50-year-olds, there are much larger differences in gene expression between twins. Few fall perfectly along the line. This process is known as genetic drift. It is thought that these differences in methylation might not be happening on random genes. Rather, certain genes might be associated with aging specifically. It is thought that these specific genes can represent an internal clock which measures how long we are alive—this is called the epigenetic clock.
Epigenetic Clock
The term epigenetic clock refers to the use of methylation to measure tissue age. The relationship between methylation and aging was first observed in spawning humpbacked salmon and was later observed in other species (Berdyshev, Korotaev, Boiarskikh, & Vaniushin, 1967). The epigenetic clock has been a topic of hot debate, and there are different ideas regarding which genes are actually involved. Models have ranged from studying only three genes (Weidner et al., 2014) to nearly 600 genes (Teschendorff et al., 2010) to make an accurate clock. Theoretically, studying more genes would be more accurate but more complicated, and studying fewer genes can be cheaper and quicker, at the expense of accuracy. Therefore, we need to find a balance between feasibility and validity. Presently, the literature best supports Horvath’s epigenetic clock, which averages methylation across 353 CpG sites (Horvath, 2013).
How do we measure methylation? The same way our cells do! When copies of DNA are made in cells, the correct parts of the DNA need to be methylated. Remember that when a gene is methylated, there are just methyl groups attached to certain cytosine nucleic acids. When the DNA is copied, it cannot just copy the DNA sequence—it needs to find a way to identify the methylated cytosines and to mark them so that they can be methylated again. If your cell just copied the DNA, without copying the methylation, then your muscle cell might divide into two completely different cells. The cell is able to remember which genes are methylated by converting the unmethylated cytosines to uracil, a nucleic base that is not naturally in DNA. This way, the cell can distinguish a methylated and unmethylated cytosine.
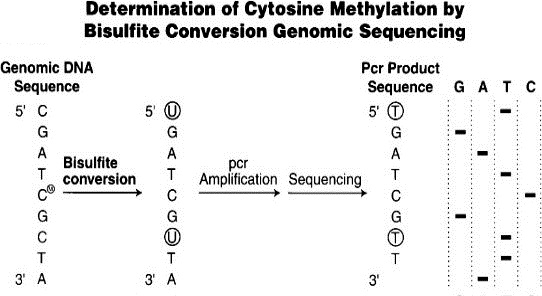
Figure 3. Bisulfite conversion enables differential sequencing of methylated and unmethylated cytosine (Weisenberger, den Berg, Pan, Berman, & Laird, 2008).
To measure methylation, we take a sample of any cell tissue, besides breast tissue, and extract the DNA. We then are able to mark the specific genes of interest. The unmethylated cytosine is then converted to uracil by adding bisulfate. The methylated cytosine bases are still cytosine, and the unmethylated cytosines are a different nucleotide altogether, which make them much easier to distinguish. We then identify which genes still contain cytosine and assume that these genes have been methylated. The number of methylated genes is then input into a model to calculate the epigenetic age. This epigenetic age—the age of your cells derived from the number of methylated genes—is generally related to chronological age—the age on the calendar year. Major takeaway–in order to figure out how aging is happening in our cells, we copy what happens in our cells naturally and do this in the lab.
Although we know that we can use epigenetics as a measure of biological age, there is still another question–how do we use this information? How many genes do we need? How specific should we be? Do we look at all the genes (and therefore spend a lot of money) or just sample a few? Scientists have been debating these questions and trying to find a reliable and accurate method for using epigenetics to measure biological age. In the next article, we’ll go over Horvath’s most recent model of the epigenetic clock and how epigenetic measures of aging have been used to assess health outcomes.
Works Cited
Berdyshev, G. D., Korotaev, G. K., Boiarskikh, G. V., & Vaniushin, B. F. (1967). Nucleotide
composition of DNA and RNA from somatic tissues of humpback and its changes during spawning. Biokhimii͡a (Moscow, Russia), 32(5), 988.
Berger, S. L., Kouzarides, T., Shiekhattar, R., & Shilatifard, A. (2009). An operational definition
of epigenetics. Genes & development, 23(7), 781-783. doi: 10.1101/gad.1787609
Fraga, M. F., Ballestar, E., Paz, M. F., Ropero, S., Setien, F., Ballestar, M. L., … & Boix-
Chornet, M. (2005). Epigenetic differences arise during the lifetime of monozygotic twins. Proceedings of the National Academy of Sciences of the United States of America, 102(30), 10604-10609. doi: 10.1073/pnas.0500398102
Horvath, S. (2013). DNA methylation age of human tissues and cell types. Genome Biology,
14(10). doi:10.1186/gb-2013-14-10-r115
Singal, R., & Ginder, G. D. (1999). DNA methylation. Blood, 93(12), 4059-4070.
Teschendorff, A. E., Menon, U., Gentry-Maharaj, A., Ramus, S. J., Weisenberger, D. J., Shen,H., … & Savage, D. A. (2010). Age-dependent DNA methylation of genes that are suppressed in stem cells is a hallmark of cancer. Genome research, 20(4), 440-446. doi: 10.1101/gr.103606.109
Weidner, C. I., Lin, Q., Koch, C. M., Eisele, L., Beier, F., Ziegler, P., … & Zenke, M. (2014).
Aging of blood can be tracked by DNA methylation changes at just three CpG sites. Genome Biology, 15(2), 1. doi: 10.1186/gb-2014-15-2-r24
Weisenberger D, den Berg D, Pan F, Berman B, Laird P: Comprehensive DNA methylation
analysis on the Illumina Infinium assay platform. Technical report Illumina, Inc, San Diego 2008.